Co robić, jeśli pacjentka nie wyraża zgody na badania? !!!
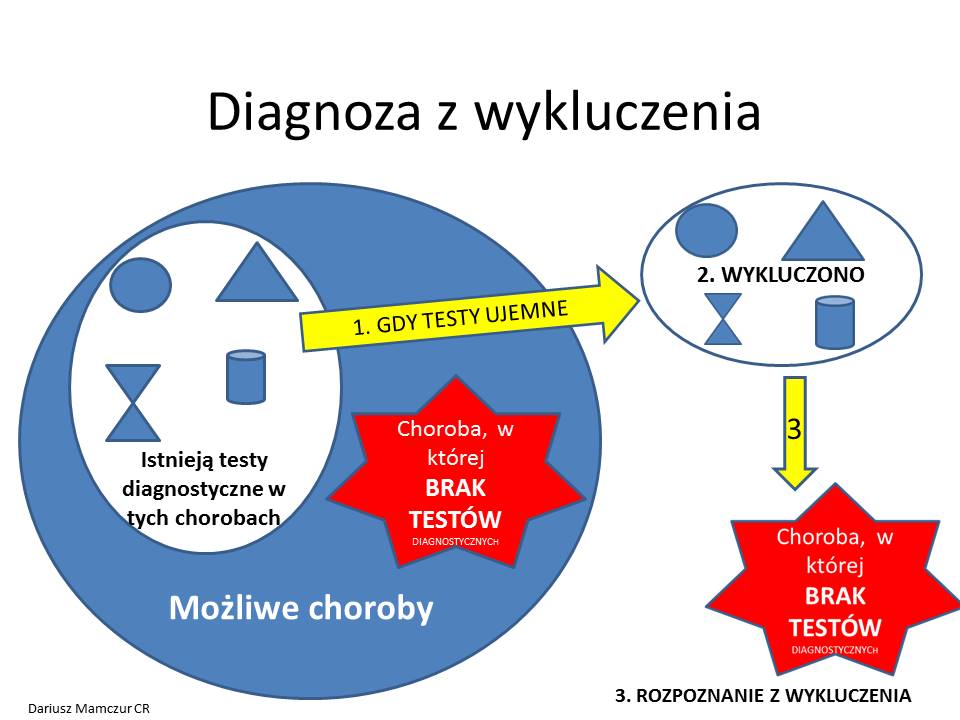
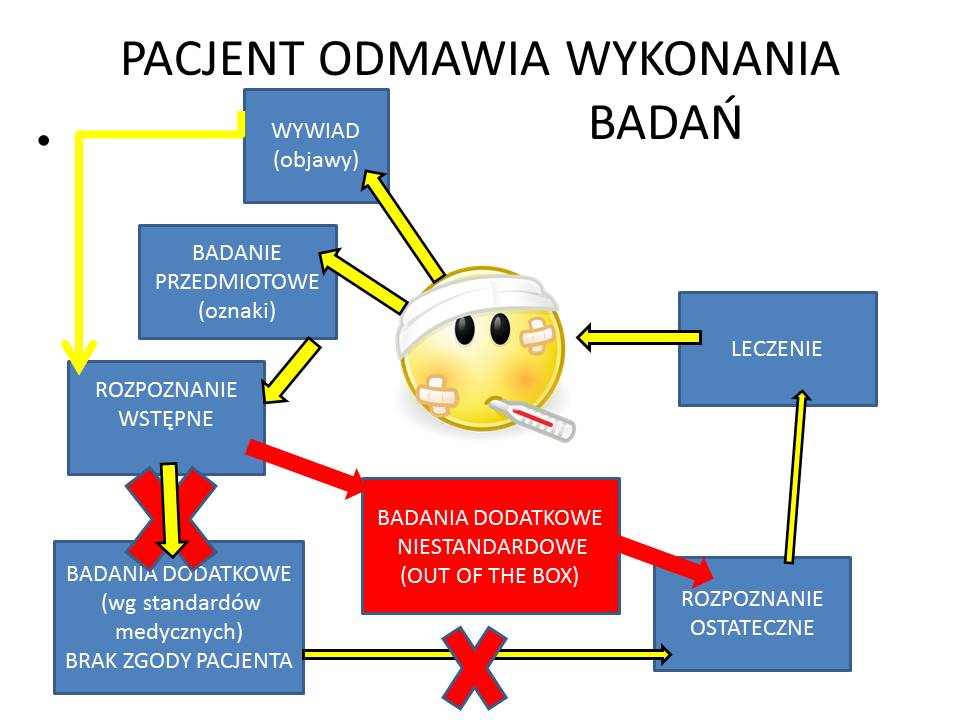
Czasem w naszym procesie diagnostycznym trudności pojawiają się ze strony samego pacjenta. Mimo tego, że mamy duże prawdopodobieństwo prawidłowo postawionej diagnozy wstępnej, dysponujemy odpowiednią bazą diagnostyczną, to jednak nie możemy potwierdzić tego rozpoznania w stu procentach. Przyczyną takiej sytuacji (oprócz standardowych przeciwwskazań do danej diagnostyki), jest czasami brak zgody pacjenta lub jego rodziny na wykonanie badań dodatkowych. W niektórych przypadkach uniemożliwia to postawienie jednoznacznego rozpoznania ostatecznego, a to może opóźnić lub wykluczyć właściwe leczenie. Ma to szczególnie miejsce w sytuacji jeśli mamy do czynienia z chorym w ciężkim stanie, a jedyną metodą leczenia jest zabieg operacyjny (np. torakotomia, kraniotomia). Wykonanie takiego zabiegu bez badań dodatkowych (nawet pod presją czasu i stanu pacjenta) naraża lekarza na odpowiedzialność prawną lekarza, w przypadku, gdyby diagnoza wstępna okazałaby się mylna. Wówczas prokurator (ale i lekarz w swoim sumieniu) stawia swoje standardowe pytanie: „Czy wykonałem właściwe czynności?” – tj. wybrałem właściwe leczenie, i „Czy wykonałem je we właściwy sposób?”- tj. zgodnie ze sztuką lekarską, standardami medycznymi, we właściwym czasie i miejscu.
Niedawno widziałem w Internecie amerykański plakat z wizerunkiem rozgoryczonego lekarza zaraz po operacji, który mówił: „Pacjenci po udanej operacji mówią, że się ona powiodła „dzięki Bogu”, ale jeśli się nie uda to MNIE pozywają do sądu!” (Wystarczy zajrzeć do narastającej w Polsce liczby toczących się spraw sądowych o błędy w sztuce lekarskiej – także tych osób z pierwszych stron gazet, pochłaniających dużo czasu, pieniędzy i emocji oraz chęci zemsty, czy wzbogacenia się). Niektóre pozwy są słuszne, inne wymagają kilkunastu biegłych (setek tysięcy złotych za ekspertyzy) i tak nie dają jednoznacznego rozstrzygnięcia wg pacjentów i ich rodzin, czy ktoś jest winny, czy nie.
Inspiracją do wpisu był film w telewizji TLC z 6 października 2016 „Ostry dyżur. Niezwykłe przypadki” (Untold stories of the ER)
 Źródło telewizja TLC Ostry dyżur. Niezwykłe przypadki
Źródło telewizja TLC Ostry dyżur. Niezwykłe przypadki
Młoda ciężarna – 38 tydzień ciąży trafiła do szpitala na ostry dyżur z powodu silnego (dysymulowanego w obecności męża) bólu w klatce piersiowej (słabsze bóle występowały od początku ciąży – chora zażywała leki gastryczne). Siła bólu 9/10 w skali graficznej wskazana przez pacjentkę. (Skala od  przez
przez  do
do  !!). Duszność oddechowa – częstość oddechów do 28/’ tachykardia do 120/’
!!). Duszność oddechowa – częstość oddechów do 28/’ tachykardia do 120/’
Pacjentce cały czas przy łóżku towarzyszył mąż (negatywnie nastawiony do służby zdrowia ze względu na dawno temu występujące powikłania u płodu w jego rodzinie po teratogennym leku talidomid).
Lekarz dyżurny Michael Gronovsky (może polskie korzenie?) wykluczył zawał mięśnia sercowego, stwierdził podwyższone D-dimery. Z wielkimi oporami uzyskał zgodę męża i pacjentki na zdjęcie rentgenowskie klatki piersiowej. Badanie wykazało poszerzenie śródpiersia górnego. Lekarz kierujący się zasadą, że „w medycynie ratunkowej ciągle modyfikuje się diagnozę w miarę napływu nowych danych”, rozważał zatorowość płucną lub tętniak rozwarstwiający aorty wstępującej. Zdecydował się wykonać tomografię komputerową klatki piersiowej. Niestety, mąż pacjentki w obawie o zdrowie płodu, zdecydowanie zabraniał żonie wyrażenia zgody na badanie. Sytuacja z minuty na minutę stawała się coraz bardziej dramatyczna, stan chorej pogarszał się. Diagnostyka zablokowana, nie można było włączyć celowanego leczenia. Cała odpowiedzialność spoczywa na lekarzu. W międzyczasie lekarz wyszedł do pokoju obok, skąd obserwował chorą. W tym czasie do łóżka pacjentki podeszła z dokumentami pielęgniarka, która przestawiła lampkę przy łóżku chorej tak, że światło padało jej na gałki oczne. Wtedy lekarz zauważył odblask ze źrenic pacjentki, który określił, że oczy wyglądały jak w filmach grozy. Zorientował się, że chora może mieć implanty soczewek. Pacjentka potwierdziła ten fakt, że wszczepiono je ze względu na podwichnięcia soczewek w dzieciństwie. Lekarz powiązał dolegliwości w klatce piersiowej i okulistyczne z możliwością choroby tkanki łącznej objawiającej się m in. tętniakami rozwarstwiającymi aorty.
(Zobacz rtg na portal porady medyczne.com.pl
W filmie nie podano nazwy choroby, jednak większość danych wskazuje, że chodziło mu o zespół Marfana (rzadka choroba częstość 10-20 na 10 000)
To w tej chorobie „w 1956 określono podobieństwo strukturalne między więzadełkami soczewek oczu a warstwą środkową aorty”, do objawów należą m. in, dziedziczone autosomalnie zwichnięcie soczewki, (link podwichnięcie soczewek)
tętniak aorty wstępującej,
Zdecydował się poprosić o interwencję kardiochirurga, który jednak nie mając pewnej diagnozy odmówił wykonania zabiegu bez wyniku badania tomografii komputerowej aorty. Lekarz znalazł się w patowej sytuacji – jeśli ma rację, nie uratuje pacjentki, wówczas grozi mu także odpowiedzialność karna. Jeśli się myli – namawianie kardiochirurga do operacji przyniesie duże skutki uboczne, nawet zgon. Powstaje dylemat – czekać na rozwój wydarzeń lub znaleźć inne potwierdzenie rozpoznania. Wybrał drugą opcję, poza standardem. ECHO nie wykluczy zatorowości płucnej, ale może potwierdzić tętniaka aorty. Poprosił kardiochirurga o wykonanie przezprzełykowej echokardiografii. Mąż pacjentki wiedząc, że USG jest bezpieczne dla dziecka – wyraził zgodę na badanie. Badanie potwierdziło rozwarstwiającego tętniaka aorty. Wykonano zabieg operacyjny. Pacjentka urodziła zdrowe dziecko.
Wniosek – „jeśli nie można drzwiami, to trzeba wejść oknem” :). Jeśli można wykonać inne niestandardowe badanie, którym możemy także potwierdzić rozpoznanie należy skorzystać z rozwiązań nietypowych. Trzeba byś kreatywnym, “wyjść z pudełka” (out of the box) schematów myślenia – ale nie każdy to potrafi.
 źródło Microsoft Office
źródło Microsoft Office
Z własnej praktyki nagłych przypadków: a) brak stazy potrzebnej do podania zastrzyku dożylnego – zastąpiłem słuchawkami lekarskim. b) brak ssaka przy porodzie dziecka poza szpitalem – zastąpiłem wężykiem od słuchawek lekarskich i zasysaniem własnymi ustami wydzieliny z ust noworodka.
 wykorzystano clipart z Microsoft Office
wykorzystano clipart z Microsoft Office
Podsumowując przedstawiony przypadek należy ciężarnej należy złożyć wyrazy szacunku dla wnikliwości obserwacji lekarza (błysk w oczach pacjentki, ocena siły bólu brzucha i zmiany w rtg śródpiersia) , jego dogłębnej wiedzy (zespół Marfana jest rzadką chorobą z wieloma objawami, trzeba było także wiedzieć, że aorta ma podobną budową do węzadełek soczewek w oku), umiejętnością kojarzenia objawów podmiotowych (Amerykanie nazywają je objawami – symptoms) z objawami z badania przedmiotowego i badań dodatkowych (rtg klatki piersiowej) (Amerykanie – oznaki [signs]- stwierdzane obiektywnie przez lekarza), determinacją do ustalenia rozpoznania (to po części intuicja, sztuka lekarska – ale wynikająca z głębokiej wiedzy i doświadczenia), umiejętność pogodzenia braku zgody rodziny pacjenta na badania z procesem diagnostycznym (znalezienie innej, nietypowe ścieżki diagnostycznej), opanowania niesamowitych emocji towarzyszących tej sytuacji.
Jeszcze raz wyrazy wielkiego uznania dla doktora Michael’a Gronovsky’ego.