
Budujemy nowe pokolenie ekspertów i Sztuczną Inteligencję
Rozwiązanie problemu lub realizacja marzeń wymaga kilkuetapowego postępowania.
zdefiniowanie problemu-> analiza problemu – w tym zebranie odpowiedniej wiedzy i kreatywnych rozwiązań (wizualizacja graficzna i filmowa rozwiązania) – stworzenie drzewa problemów i rozwiązań oraz algorytmów dostosowanych do konkretnych sytuacji -> wdrożenie programów i projektów realizacji – stworzenie najlepszych i powtarzalnych w nowych sytuacjach list “to do”-> implementacji do sztucznej inteligencji.
Narzędzia do gromadzenia wiedzy już mamy, nad sztuczną inteligencją potrafiącą wytwarzać algorytmy i przekazać je maszynom dopiero pracujemy.
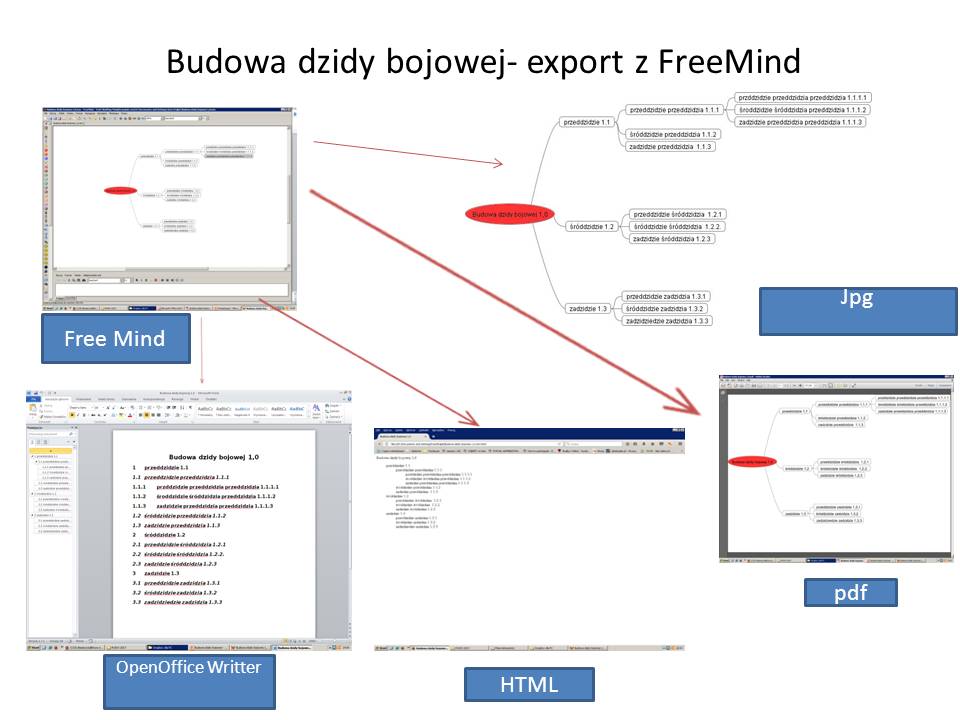
Wadą dzisiejszych baz wiedzy (drukowanych, audio, wideo oraz ich wersji elektronicznych) jest powielanie tych samych rozwiązań tylko innymi słowami. Stąd miliony wyników wyszukiwania danej frazy, które tylko w niewielkim procencie wnoszą coś oryginalnego do naszego działania. Bazy wiedzy w większości są dopiero „paliwem” do tworzenia rozwiązywania naszych wyzwań i problemów. Informacje z nich pochodzące muszą dopiero przeanalizowane, z nich stworzone sposoby i kolejność działania, aby uzyskać sukces. Całościowa (holistyczna) analiza problemu możliwa jest dzięki różnym narzędziom – w tym np. wizualizacja graficzna, burza mózgu oraz mapy myśli (to one nie pozwalają na dublowanie treści – od razu widać, czy nowa wersja mapy wnosi coś wartościowego). Zinwentaryzowanie i hierarchizacja (nadanie kodów wyszukiwania) poszczególnych map, pozwoli wykorzystywać je w tworzeniu rozwiązań dopasowanych do nieco odmiennej sytuacji. Wytworzone rozwiązanie można dodać do istniejącej globalnej mapy i tak może działać „perpetuum mobile” rozwoju wiedzy i umiejętności ludzkości (uzupełniamy mapę o dodatkowe gałęzie). Prawidłowa mapa myśli (będąca pod nadzorem i oceną internautów- nadawanie rang podniesie wiarygodność danego modelu rozwiązań. Zaletą map myśli (np. bezpłatnej FreeMind)jest możliwość wygenerowania raportów w plikach pdf, jpg, html, openOffice Writer, TWiki. Można linkować do elementów mapy, stron www i plików zewnętrznych. Program wyszukuje frazy w obrębie mapy. Ponadto można importować do mapy i wyświetlać widok zdjęć w plikach jpg (ale są one w odrębnych katalogach- nie przenoszą się razem z kopiowaniem mapy), można wklejać skopiowane fragmenty tekstu, wpisywać i przeglądać foldery z oraz wstawiać (dołączać w dowolnym miejscu mapy zewnętrzne!!! – to jest podstawa do tworzenia Globalnej Dynamicznej Mapy Myśli i Algorytmów.
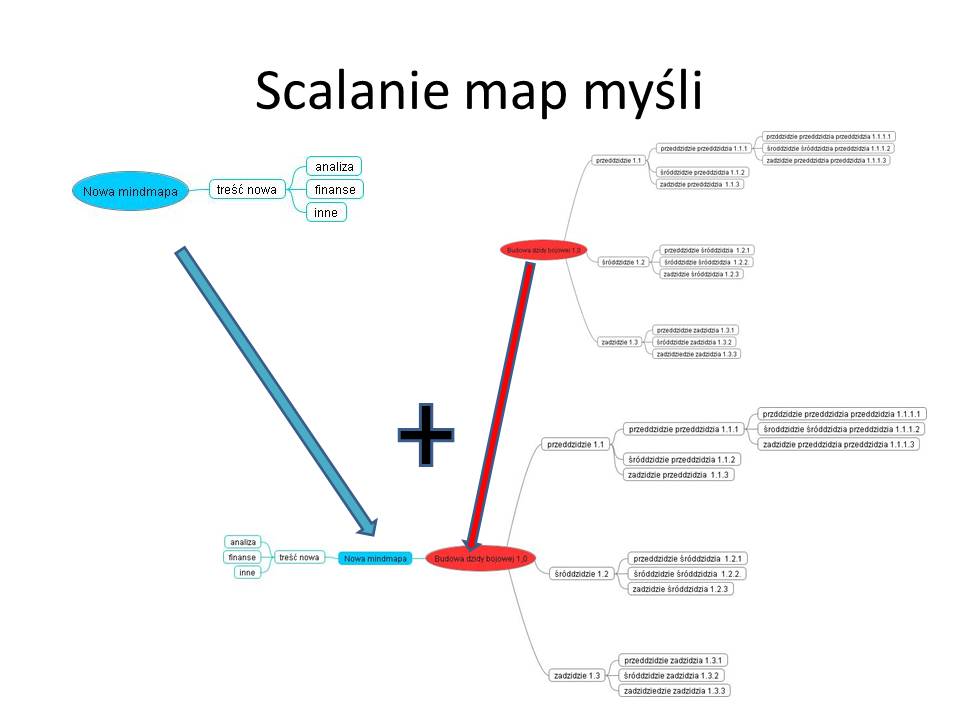
Do demonstracji tych możliwości posłużę się zabawą językową w opisywanie “dzidy bojowej” – jest to też świetny przykład tworzenia coraz bardziej rozbudowanych katalogów – a na nich chce opierać nowa przeglądarkę internetową.
Dzida składa się: z przeddzidzia, śróddzidzia i zadzidzia.
Przeddzidzie składa sie z przeddzidzia przeddzidzia, śróddziadzia przeddzidzia i zadzidzia przeddzidzia. Idąc dalej przeddzidzie przeddzidzia składa sie z przeddzidzia przeddzidzia przeddzidzia, śróddzidzia przeddzidzia przeddzidzia i zadzidzia przeddzidzia przeddzidzia. I tak dalej. 🙂
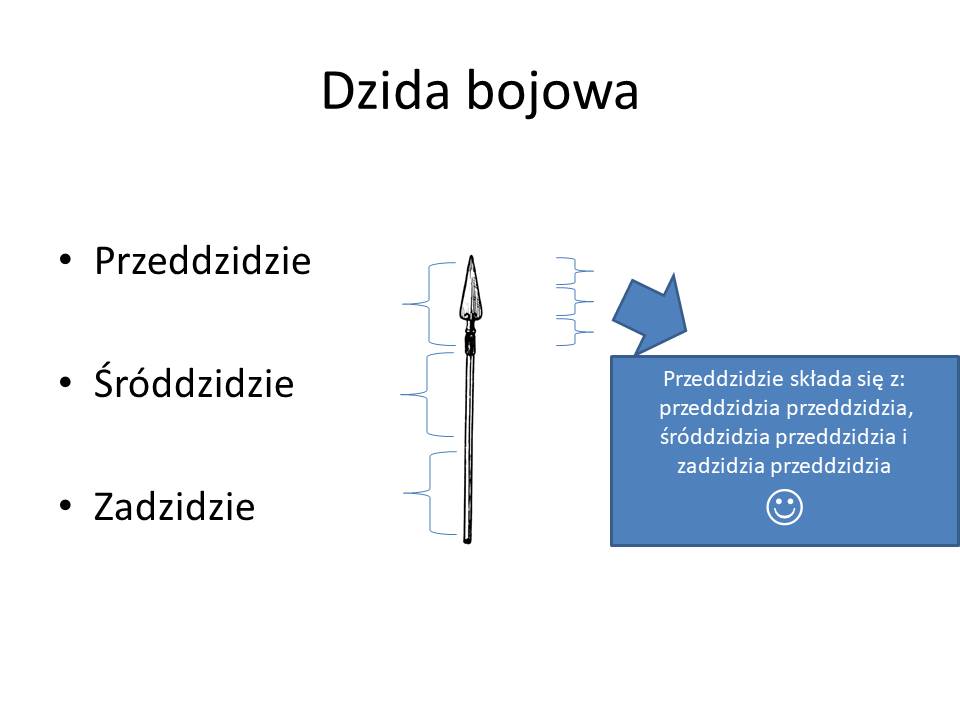 Jeśli zapiszemy to w postaci mapy myśli to otrzymamy taki obraz mapy myśli
Jeśli zapiszemy to w postaci mapy myśli to otrzymamy taki obraz mapy myśli
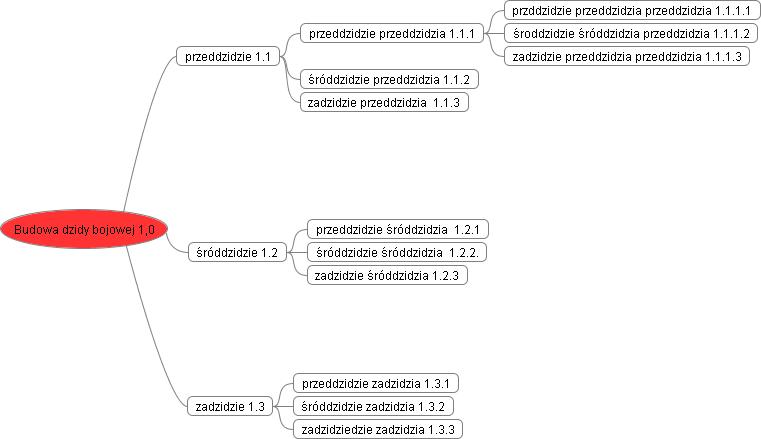
Jeśli zaimportujemy rysunek to obraz mapy może wyglądać następująco
 Poniżej pokazuję możliwości eksportu danych z mapy myśli
Poniżej pokazuję możliwości eksportu danych z mapy myśli
Na chwilą obecną może być problem z automatycznym importem do map myśli (wyszukiwanych semantycznie) i tworzeniem dodatkowych gałęzi mapy nowych informacji. Na przykład mamy opis procedury i podstawy prawnej danego zagadnienia – jak je zaktualizować bez udziału człowieka o nowe ustawy (w tym podpisane ostatniej nocy) 🙂
Mapy powinny mieć też mieć możliwość wygenerowania list protokołów staranności (must to do). Z tych map i list „to do” tworzymy algorytmy decyzyjno- wykonawcze. Algorytmy odpowiednio skodyfikowane (np. można wyjść od klasyfikacji działalności gospodarczej- lub stworzyć odrębny system), lub już używane kategorie np. przez Wikipedię – W TWiki, Media Wiki (zwróćmy uwagę na minusy stosowania w/w kategorii – mapa myśli ograniczyłaby dublowanie informacji) także będą mogły być szybko wyszukiwane i wykorzystywane w innych sytuacjach oraz w połączeniu z aktualizacją baz wiedzy, map myśli stanowić szybkie wdrażanie projektów. Jak będziemy wiedzieli co i w jakiej kolejności mamy zrobić, to pozostaje nam zdefiniowanie celów, stworzenie programów, projektów i przydzielenie zadań – ustalenie, co, kiedy i do kiedy, przez kogo, dlaczego oraz za ile ma być zrobione. Przy takich narzędziach – programach eksperckich (w informatyce zwanych ekspertowymi), każdy człowiek może stać się ekspertem w wielu dziedzinach (nawet bez wykształcenia kierunkowego – wystarczy umiejętność obsługi komputera, aby zachowując należyta staranność np. zbudować most na Wiśle. Jeśli będziemy planować nasze działania biznesowe, to już nie dojdzie do upadku tak licznej grupy firm w początkowym okresie ich działania, już nie tylko co 100-ny start-up skończyłby się sukcesem – gdzie będzie wtedy ludzkości – „kosmiczny odlot”, ochrona środowiska, oszczędności pieniędzy i zasobów. Jak się zabezpieczyć przed hakerami i brakiem zasobów do przechowywania danych – sposobem jest sieć rozproszona i kryptografia z wykorzystaniem łańcucha blokowego (blockchain).
Kto ma przeanalizować problem, stworzyć drzewa strategii i problemów, przeszukać bazy danych, stworzyć mapy myśli, algorytmy , listy „must to do” (w tym filmy instruktarzowe – np. “spryciarze.pl, storyboard= scenorys i inne) oraz plany projektów oraz wdrożyć ich realizację.
Początkowo powinien wykonywać to człowiek, ale w miarę rozwoju map w formacie „big data” będą one uzupełniane o automaty komputerowe – sztuczną inteligencję. W tej chwili rozpoczął się wyścig miedzy mocarstwami i potentatami internetowymi. Zauważono, ze sztuczna inteligencja może być motorem postępu technologicznego, naukowego ale także polityczno – militarnego. Jeśli z wyprzedzeniem będziemy wiedzieć, co w danej sytuacji zrobi konkurencja lub wróg – to odpowiednio przygotujemy nasz arsenał militarny oraz możemy wykonać uderzenie wyprzedzające na miliony wariantów, które mógłby „na piechotę”, tj. bez algorytmów, wymyślić przeciwnik. Na przykład – czy przeciwnik w danej sytuacji wystrzeli głowice atomową, gdzie ja może skierować- w czasie lotu w milisekundach oceniamy tor lotu i warianty zmiany jego toru, gdzie zniszczyć głowice z największym prawdopodobieństwem, co zrobić, jeśli obrona się nie uda – jakie procedury uruchomić na danym terenie- podobnie jest w biznesie). W to zagadnienie wpisuje się myślenie – „A co się stanie, gdy sztuczna inteligencja zaatakuje człowieka? Musimy wiedzieć, czego ją nauczyliśmy (baza map myśli i algorytmów), jaką wiedzą ona dysponuje i leczyć, ze analiza baz danych i algorytmów da człowiekowi przewagę dzięki kreatywnemu myśleniu i zaatakuje jej słabe strony, na które nie była w stanie wytworzyć swojej broni.
W chwili obecnej, gdy Google (Forbes 08/18 str. 114,116 „Sztuczna inteligencja w wyścigu po pieniądze”- Piotr Karnaszewski) zbudował system sztucznej inteligencji pozwalający wygrać z mistrzem chińskiej gry w „Go” Lee Sedola – wykonując ruchy w ciągu 0,1 sekundy ruszył wyścig. Gra GO ma ponoć więcej możliwych kombinacji niż jest atomów na ziemi. Chińczycy postawili na sztuczną inteligencję (AI) w praktyce. Wg ich założeń już 2025 sztuczna inteligencja ma być motorem napędowym chińskiego przemysłu . Szacuje się, ze za 10 lat wartość światowego rynku AI przekroczy 16 mld dolarów. „Sukcesy Chińczyków w dziedzinie badań AI biorą się z dwóch faktów: ogromnych ilości środków przeznaczonych na ten cel oraz możliwości rozwijania algorytmów. Aby zbudować sztuczna inteligencję potrzeba mnóstwa danych, a nic ich nie produkuje lepiej niż ludzie. I tu wychodzi przewaga chińskich firm technologicznych – z ich produktów korzysta aż 730 mln internautów. Dostęp do tak dużych zbiorów i wzorców zachowań z codziennego życia Chińczyków pozwala badaczom AI prowadzić badania na znacznie większą skalę i większą intensywnością niż ich zagranicznym odpowiednikom”.
Dlaczego nie wykorzystać Globalnej siły Internautów i zamienić wyszukiwarkę WWW na wyszukiwarkę wyższej generacji – GDMM i A (globalną mapę myśli i algorytmów opartej na wiedzy opisanej werbalnie ale odpowiednio przefiltrowanej pod kątem ich nowatorskiej unikatowej wiedzy i przydatności ). Stwórzmy narzędzia (np. język programowania LUNA pozwalający analizować dane w sposób graficzny z naniesionymi źródłami danych stworzony przez polskich programistów – laureatów z 2018 roku Global Impact Challenge-Wojciech Daniło i Marcin Kostrzewa) (Newsweek28.05-3.06.2018 str. 64) (niestety brak szczegółów i demo oprogramowania w internecie), społeczność oraz modele biznesowe, które pozwolą rozwijać te ideę, tak aby każdemu się opłacało tworzyć wartościowe algorytmy – firmom i indywidualnym osobom. W kontekście sztucznej inteligencji uznałem, że mój pomysł z 2011 roku o Globalnej Mapie Myśli powinienem uzupełnić o wynikającą z niej mapę Algorytmów – pozwalającą robotom szybciej implementować widzę z map myśli. Obecnie algorytmy pomagają lekarzom m.in. w diagnostyce różnicowej i stawianiu rozpoznań. Istnieją portale poświęcone algorytmom np. portal Medal (The Medical Algorithms Company – duża wyszukiwarka algorytmów i kalkulatorów medycznych),
albo też oparte na algorytmach stawianie rozpoznań Isabel(płatny) Diagnosaurus, ( był świetny portal- ale zniknął- diagnosispro i podobne) na podstawie objawów łączące algorytmy kilku objawów z prawdopodobieństwem występowania chorób oraz zaleceniami co do wykonania badań dodatkowych, które mają potwierdzić rozpoznanie. Niestety, algorytmy różnych firm mogą opierać się na innych danych – nie są porównywalne i wystandaryzowane. Mapa myśli mogłaby to uporządkować.
GDMMiA byłaby walką ze śmieciami znajdującymi się w big data. Nową wyszukiwarką -nie tylko wiedzy ale też praktycznych rozwiązań i planów działania w poszczególnych sytuacjach. Baza do robotyzacji i AI. Te moce obliczeniowe mogłyby być wykorzystane bardziej produktywnie. Ile jest obecnie w Internecie kursów , książek, filmów pseudoekspertów, które nie wnoszą nic nowego, na ile sposobów ten sam autor sprzedaje te same rozwiązania w kolejnych książkach, ubierając je tylko w inne słowa – a my tracimy czas na ich bezproduktywne czytanie (to jak stacje TV- sprzedają te same wiadomości ale z innym komentatorem) .
Sztuczna inteligencja interesuje największe firmy technologiczne. Także Apple – obecny „smok” w dziedzinie biznesowej (smok to przychód ponad 1 bilion= 1012 Dolarów (jednorożec to miliard 109) także wdraża sztuczną inteligencję upatrując w niej szanse na walkę na rozwój (nie chce być kolosem na glinianych nogach).
W chwili obecnej sztuczna inteligencja wykorzystywana jest przez Facebooka do optymalizacji reklamy, w handlu, przez chiński odpowiednik Google-Baidu do autonomicznego kierowania autobusem, w medycynie – f-ma Tencenta do wykrywania raka płuc (przeszukuje w kilka sekund bazę 300 tys. zdjęć rentgenowskich chorych z rakiem płuc i w kilka sekund porównując ze zdjęciami płuc pacjenta stawia rozpoznanie czy badany chory ma raka płuc. Podobne narzędzie do porównywania obrazu chorób skóry (jest ich 2 tysiące – a doświadczony dermatolog w swoim zżyci widział mniej niż 800) ze zmianami na skórze pacjenta. Właściwe rozpoznanie, to prawidłowe, skuteczne i szybkie leczenie. Dr Watson – mój wpis na blogu z 2.12.2016 r – Kiedyś Lem, dzisiaj sztuczna inteligencja – IBM Watson
Co zrobić, aby ludzie chcieli tworzyć GDMMiA? Ludzi motywuje sława, władza, seks i pieniądze. W tym przypadku z pomocą może przyjść kryptograficzna technologia blockchain oraz oparte na niej kryptowaluty np. bitcon. ( W Polsce tą dziedziną pasjonuje się łódzka firma BinarApss a w niej wiceprezes Maciej Krasowski– 10 września 2018 odbędzie się druga edycja meetupu “Blockchain Business Łódź” zrzeszającego fanów #blockchain i kryptowalut). Pozwala ona uszeregować wpisy w łańcuchu, których nie można zmienić (pewien rodzaj urzędu patentowego – i prawo do sławy tego, kto wprowadził pierwszy dane rozwiązanie), ma rozproszoną i kryptograficznie zabezpieczoną strukturę danych (trudną ją zaatakować hakerom) – czyli duża stabilność i oporność na przeciwności losu, można płacić za usługę stworzenia pewnego fragmentu mapy bezpiecznie i bez pośrednictwa banków. Być może wyłączy konkretnego administratora danych i pozwoli na rangowanie wartości algorytmów automatycznie przez społeczność internetową -“wykopiemy” najlepsze listy staranności i “to do”.
Optymalne byłoby importowanie do map myśli już wykonanych map ale prezentowanych w plikach graficznych – jpg, tif, powerpoint, filmowych, zdjęć ze smartfonów – pozwoliłoby to na automatyczne, małym kosztem wstępne i szybkie stworzenie GDMMiA (np. komputer dr Watson analizuje zdjęcia medyczne). Nie znam programu, który może wykonać tę funkcję, a potem połączonego z robotem przeszukującego wszystkie strony www (tak jak to robi z danymi tekstowymi Google, który skopiował i skatalogował już chyba cały Internet. Mapy myśli i algorytmów powinny się spotkać w jak najszybszym czasie uzupełniać z superinteligencja reprezentowana przez sztuczną inteligencję (sztuczny mózg, komputerowe AI, implanty domózgowe, sieci neuronowe itd.). Superinteligencja przewyższy wielokrotnie nasza sztuczną inteligencje – bo ma być wielokrotnie mądrzejsza od tego co wymyśli pojedynczy człowiek i cała ludzkość!!!
Jak zawiadywać tak olbrzymią ilością informacji, sprzęgać wizualizację graficzną z pokładami wiedzy spisanymi na stronach www – do tego przyda się wiedza i oprogramowanie dużych baz danych, czyli biga data (teraz korzysta z tego wyrafinowana korporacyjna reklama).
Przydatne będą też wykorzystywane w business intelligence: eksploracja danych , eksploracja procesów , sieci neuronowe, systemy ekspertowe oraz algorytmy genetyczne ,
Na to wszystko czekałem w moim projekcie od 2011 roku. 🙂
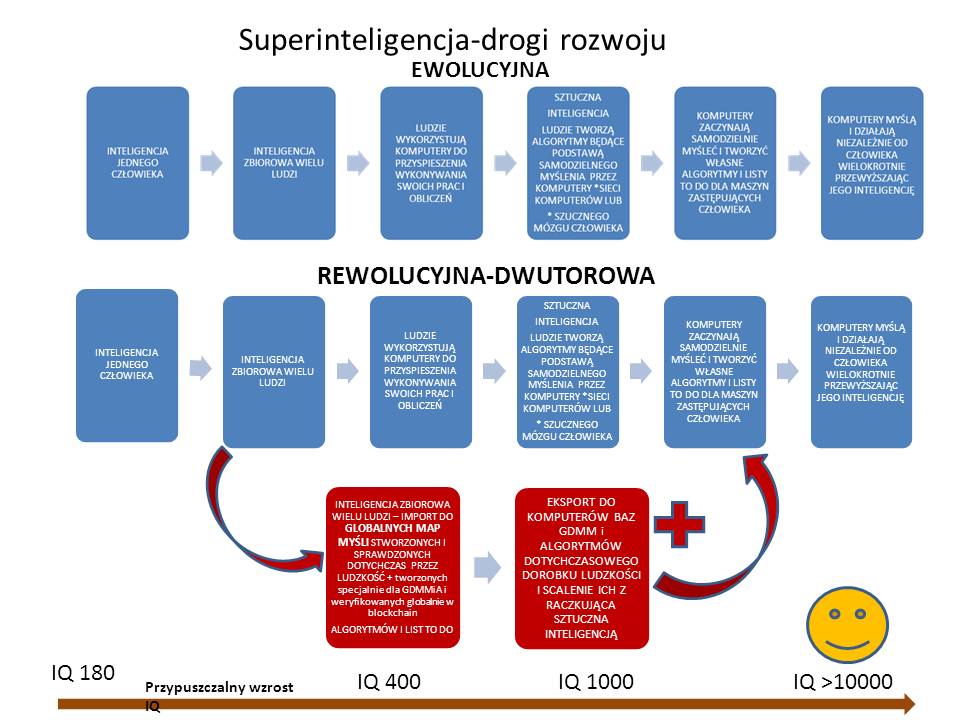
W czym GDMM jest lepsza na tym etapie od tworzonej superinteligencji (tj. inteligencji wyższej ni ludzka i opartej na samodzielnej analizie i wnioskowaniu komputerów? (AI)
Moim zdaniem superinteligencja chce przeskoczyć pewien etap ewolucji. Nasza ludzka wiedza powstaje ewolucyjnie dzięki kumulacji doświadczeń przez kolejne pokolenia. Nie można zacząć biegać zanim nie zaczniemy raczkować. Proponuję zatem etap pośredni, gdzie to początkowo ludzie będą sami tworzyć rozwiązania algorytmiczne poszczególnych rozwiązań drobnych problemów poprzez ich dogłębną analizę i weryfikację w konkretnych sytuacjach (ewolucyjnie słabsze wersje będą wypierane przez lepsze). Metoda jest prosta, narzędzia są już dostępne, trzeba tylko zbudować bezpieczny, globalny system do rozwijania, weryfikowania i przechowywania informacji. Jak stworzymy mapę niepowtarzalnych algorytmów (nazywam je cegiełkami mapy myśli), dajmy w ten sposób abecadło komputerom, aby stopniowo tworzyły pod kontrolą ludzi proste algorytmy. Dopiero na bazie algorytmów i map myśli zacznijmy tworzyć superinteligencję, która mając już “zaimplementowane odruchy bezwarunkowe (tak jak człowiek robi pewne rzeczy nieświadomie), będzie budować z tych “cegiełek map myśli” nowe konstelacje rozwiązań, a na bieżąco ludzie “na piechotę” i tak będą mogli od razu implementować te rozwiązania. Sprzedając własne mapy myśli z algorytmami, będą mieli motywację do tworzenia globalnej społeczności organicznie budującej GDMMiA oraz szybszy rozwój ludzkości. Na razie wyrafinowane technologie zostawmy naukowcom, a wykorzystajmy potencjał mózgów ludzi całego świata. Tutaj Chiny, czy Indie a także USA i Rosja mają olbrzymie możliwości organizacyjne, aby takie rozwiązanie wprowadzić.
W osobnych wpisach pokażę jak zaczynałem myślenie o GDMM (potem GDMMiA) wykorzystując moją wiedzę i potrzeby w automatyzacji diagnostyki trudnych przypadków chorobowych. Zainspirowała mnie one do głębszej analizy problemu wnioskowania i tworzenia nowych rozwiązań. Nie tylko Sherlock Holmes zaczerpnął myślenie z medycyny, także informatycy mogę na tym dużo skorzystać. 🙂
Proponuję też przeczytać 2 książki, które właśnie kupiłem:
Nick Bostrom – Superinteligencja. Scenariusze, strategie, zagrożenia. Helion-2016 . Oryginał z 2014 – ale już nieaktualne informacje na temat gry GO – (opisałem powyżej – komputer wygrał z graczem 1-go dana!!!) – autor – szwedzki profesor, profesor Uniwersytetu Oksfordzkiego, kierownik Instytutu Przyszłości Ludzkości działającego w ramach Oxford Martin School
oraz
Tom Griffiths i Brian Christian – Algorytmy, kiedy mniej myśleć i inne sposoby na
racjonalne życie. Wydawnictwo JK 2018
Mapy myśli jako narzędzie burzy mózgów i współpracy w chmurze opisywałem już na blogu
O blockchain w medycynie opowiada John Sotos – konsultant medyczny filmu Doktor House” w filmie Intel’s John Sotos – Distributed: Health 2017

