Temat omawiam na podstawie artykułu w Medical Tribune nr 5 maj 2018 str 54-57-„Noworodek z wrodzoną wadą metabolizmu”- Monika Stelmach rozmawia z dr hab. prof. nadzw. PUM Marią Giżewską z Kliniki pediatrii, Endokrynologii, Diabetologii Chorób Metabolicznych i Kardiologii Wieku Rozwojowego PUM oraz artykuł także w Medical Tribune „Odyseja diagnostyczna” str. 50-53 – wywiad Anny Hucko z dr hab. prof. nadzw. Jolantą Sykut-Cegielską, konsultantem krajowym w dziedzinie pediatrii metabolicznej, kierownikiem Kliniki Wrodzonych Wad Metabolizmu i Pediatrii IMiD w Warszawie.

„Wrodzone wady metabolizmu (WWM) to uwarunkowane mutacjami pojedynczych genów zaburzenia w przebiegu procesów biochemicznych zachodzących w ustroju. Definicja ta odróżnia WWM od schorzeń metabolicznych uwarunkowanych wielogenowo lub środowiskowo, takich jak cukrzyca czy hiperlipidemia”. Pojawiają się one rzadko, czyli nie częściej niż 5 razy na 10 tysięcy urodzeń.
Klasyfikacja WWM (według prof. Jean-Marie Saudubraya):
- choroby, które przebiegają z objawami zespołu zatrucia (intoksykacji),
- choroby z zaburzeniami procesów energetycznych,
- choroby z zaburzeniami metabolizmu dużych molekuł
Klasyfikacja WWM z uwzględnieniem potencjalnych możliwości terapeutycznych w stanach nagłych (Wg PREITSCH v. ET Al.
| 1. WWM PRZEBIEGAJĄCE Z OBJAWAMI ZESPOŁU ZATRUCIA NP.: |
| · Zaburzenia cyklu mocznikowego,
· Aminoacidopatie: tyrozynemia, choroba syropu klonowego, fenyloketonuria
· Wrodzone kwasice organiczne
· Niektóre zaburzenia b-oksydacji kwasów tłuszczowych, np. deficyt LCHAD,
· Zaburzenia metabolizmu cukrów: galaktozemia, wrodzona nietolerancja fruktozy |
| 2. WWM Z OBNIŻONĄ TOLERANCJĄ NA GŁODZENIE, NP.: |
| · Glikogenozy
· Zaburzenia glukoneogenezy,
· Zaburzenia b-oksydacji kwasów tłuszczowych,
· Zaburzenia ketogenezy i ketolizy
· Deficyt transportera karnityny,
· Hiperinsulizm |
| 3. WWM Z ZABURZENIAMI WEWNĄTRZMITOCHONDRIALNEGO TWORZENIA ENERGII, NP.: |
| · Niedobór kompleksu dehydrogenazy pirogronianu,
· Zaburzenia czynności łańcuch oddechowego |
| 4. WWM Z ZABURZENIAMI NEUROTRANSMISJI, NP.: |
| · Drgawki pirydoksyno (witamino-B6) zależne,
· Drgawki odpowiadające na fosforan pirydoksyny lub kwas foliowy |
| 5. WWM Z OGRANICZONYMI MOŻLIWOŚCIAMI TERAPEUTYCZNYMI W SANACH NAGŁYCH DEKOMPENSACJI METABOLICZNEJ, NP.: |
| · Nietoksyczna hiperglicynemia,
· Deficyt oksydazy siarczynowej,
· Inne (poza typem A) formy deficytu kofaktora molibdenowego |
Kiedy podejrzewać WWM:
Wywiad rodzinny:
– zgony dzieci w rodzinie z podobnymi objawami lub zgony o nieustalonej etiologii
– w rodzinie epizody ALTE (ostre epizody zagrażające życiu)
– występowanie u dziecka jakichkolwiek zaburzeń napadowych (niekoniecznie padaczkowych- w tym lekoopornych)
– opóźniony rozwój dziecka bez jasnej przyczyny
Dane z wywiadu i badania przedmiotowego nie są specyficzne, nie można zidentyfikować jednego flagowego objawu (np. makrocefalia może, ale nie musi być WWM)
Najczęstsze błędne rozpoznania zamiast WWM to:
- sepsa (gdy objaw kliniczny ją przypomina lub jest objawem intoksykacji)
- Mózgowe porażenie dziecięce w postaci wiotkiej lub dystonicznej w przypadku dzieci bez żadnego obciążenie okołoporodowego, które uzyskały 10 punktów w skali Apgar (wiotkość i dystonia są objawami wielu wrodzonych wad metabolizmu związanymi np. z zaburzeniami mitochondrialnymi lub neurotransmiterowymi.
- Pylorostenoza – u dzieci wymiotujących
- Wrodzona wada serca – dzieci męczą się przy jedzeniu
- Krytycznie powinno się też oceniać rozpoznania zakażenia wewnątrzmacicznego lub urazu okołoporodowego – o ile nie są one potwierdzone
Sytuacje prowokujące ujawnienie się choroby:
- Nawet niegroźne infekcje
- Dekompensacja kliniczna w przedłużonym głodzeniu. Np. przy biegunce rotawirusowej, zapaleniu ucha, anginie (tak sytuacja może być zagrożeniem życia u dziecka np. z acydurią metylomalonową lub deficytem MCAD, lub hiperamonemią pierwotną)
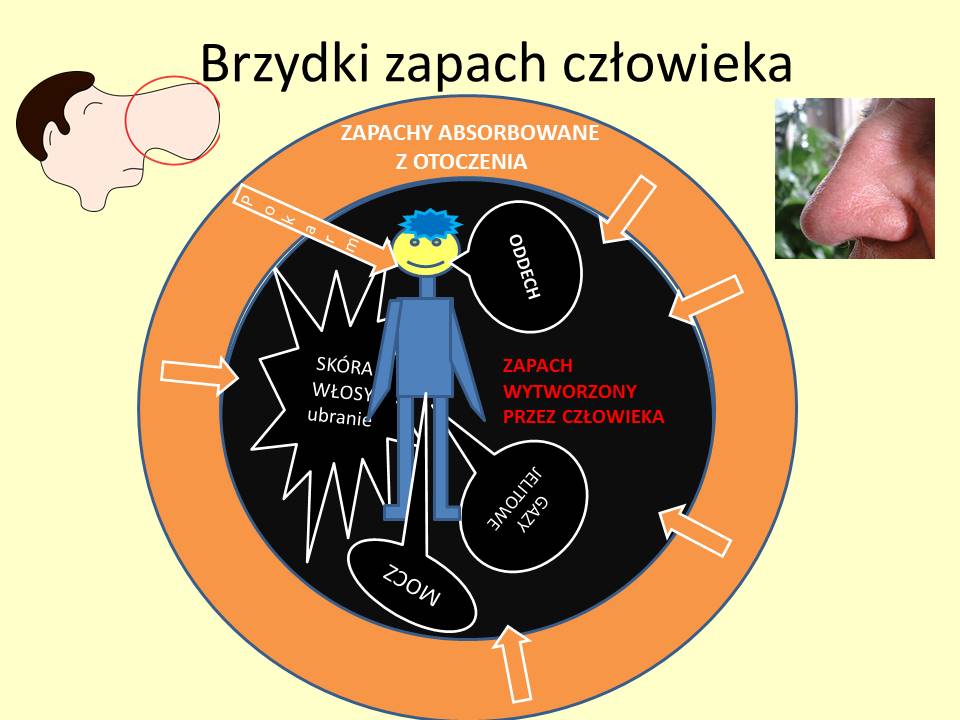
Specyficzny zapach pacjentów z WWM
| WRODZONY BŁĄD METABOLIZMU – ZAPACH MOCZU |
| Kwasica glutarowa t. II – spoconych nóg |
| Choroba syropu klonowego – syropu klonowego |
| Fenyloketonuria – stęchły mysi |
| Tyrozynemia – kapuściany, zjełczały |
| Cystynuria – siarki |
| 3-metylokrotonyloglicynuria deficyt wielu karboksylaz- moczu kocura |
Badania przesiewowe WWM
- Badania podstawowe:
- Morfologia krwi ( możliwa niedokrwistość, trombocytopenia, leukopenia)
- Gospodarka kwasowo-zasadowa (podwyższenie luki anionowej w acyduriach alkaloza oddechowa w hiperamonemiach),
- Stężenie glukozy (możliwa zaró1.no hipo- jak i hiperglikemia),
- jonogram w surowicy krwi (hipokacemia),
- podwyższone stężenie amoniaku (ważny czas od pobrania krwi do przeprowadzenia oznaczenia),
- podwyższone stężenie kwasu mlekowego (zakażenie, niedotlenienie, pobranie krwi w niepokoju dziecka mogą dać istotne podwyższenie kwasu mlekowego),
- badanie funkcji wątroby,
- badanie ogólne moczu (ketonuria w acyduriach organicznych, objawy uszkodzenia cewek proksymalnych – zespół Fanconi-de Toni-Debre w galaktozemii, tyrozynemii, fruktozemii, o ile do diety w późniejszym wieku dołączona jest fruktoza).
2.Badania specjalistyczne w kierunku WWM:
- profil acylokarnityn w suchej kropli krwi metodą MS/MS (tandemowa spektrometria mas)
- profil kwasów organicznych w moczu metodą chromatografii gazowej sprzężonej z spektrometrią mas (GS-MS),
- aminogram osocza i płynu mózgowo-rdzeniowego,
- stężenie neurotransmiterów w płynie mózgowo-rdzeniowym,
- inne – w zależności od potrzeb
- Badania genetyczne: kariotyp, inne:
4.Badania po śmierci dziecka
Po zgonie pobrać:
- mocz GC MS (jak najwięcej)
- surowicę, osocze (2-2,5 ml)
- płyn mózgowo-rdzeniowy (0,5-1ml)
- kroplę krwi i żółci na bibułę do badań przesiewowych
- czasami fragment skóry do hodowli fibroblastów
- krew pełną (ok. 10 ml) do ewentualnej izolacji DNA
Ustalenie przyczyny śmierci dziecka pozwoli rodzinie świadomie zdecydować o prokreacji w przyszłości.
Badanie suchej kropli krwi metodą tandemowej spektrometrii mas pozwala na podejrzenie 26 wrodzonych wad metabolizmu spośród ponad tysiąca istniejących wrodzonych wad metabolizmu.